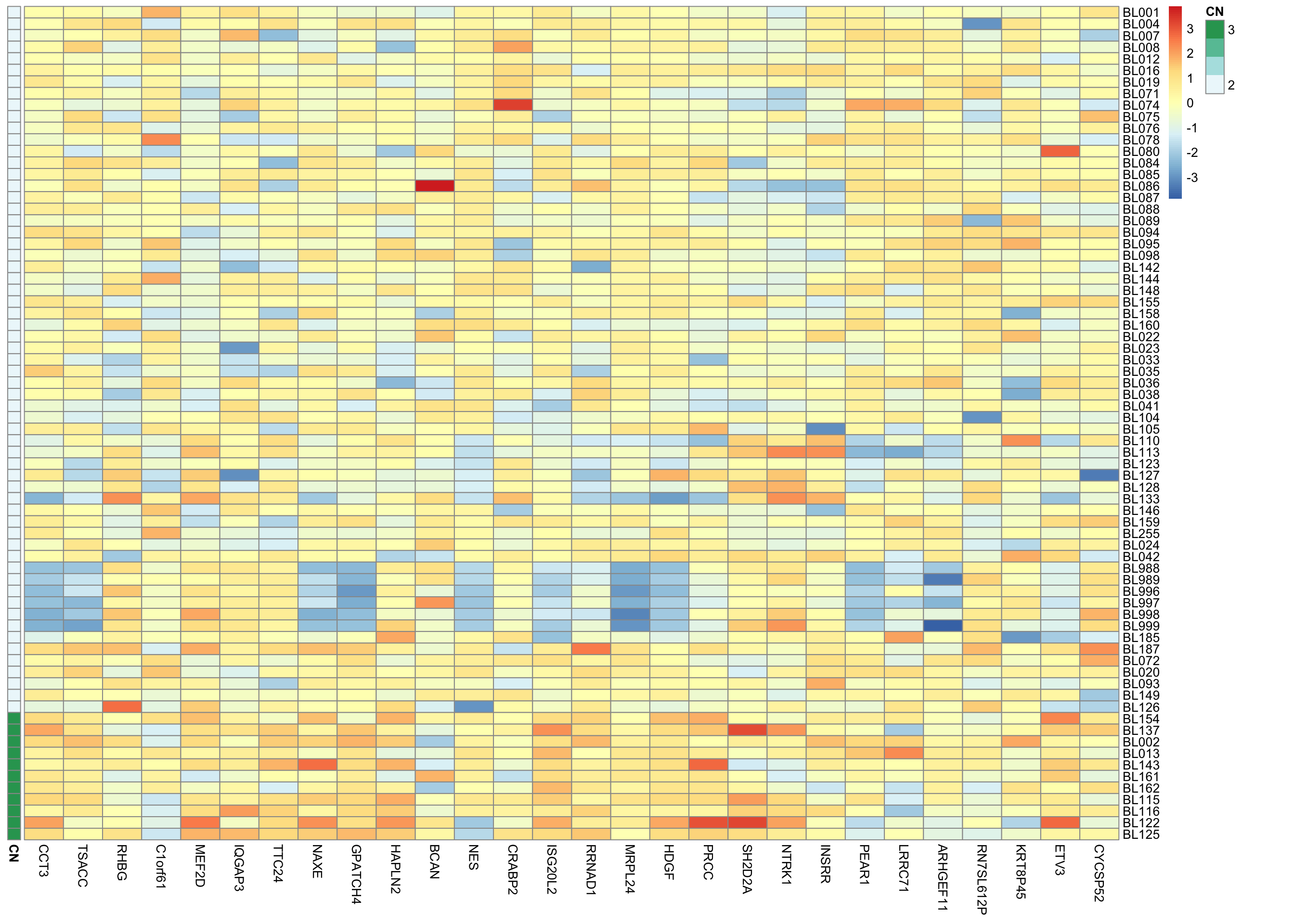
plot_region_expr <- function(chrom, start.pos, end.pos, biomart, salmon, sequenza_df, ...) {
genes <-
getBM(
attributes = c("chromosome_name", "start_position", "end_position", "hgnc_symbol"),
filters = c("chromosome_name", "start", "end"),
values = list(chromosome_name = sub("^chr", "", chrom), start = start.pos, end = end.pos),
mart = biomart) %>%
subset(.$hgnc_symbol %in% rownames(salmon$clean$counts)) %>%
arrange(start_position) %$%
hgnc_symbol
annot <-
sequenza_df %>%
filter(chromosome == chrom, end > as.numeric(start.pos), start < as.numeric(end.pos)) %>%
group_by(sample) %>%
summarise(segmean = mean(segmean) + 2) %>%
mutate(patient = get_patient_id(sample)) %>%
select(patient, CN = segmean) %>%
as.data.frame() %>%
column_to_rownames("patient")
cbs <- setdiff(colnames(salmon$clean$cvst), rownames(sequenza_df))
annot <- rbind(annot, data.frame(CN = rep(2, length(cbs)), row.names = cbs))
patient_order <- annot %>%
rownames_to_column("patient") %>%
arrange(CN) %$%
patient %>%
intersect(colnames(salmon$clean$counts))
pheatmap::pheatmap(t(assay(salmon$clean$cvst)[genes, patient_order]),
cluster_cols = FALSE, cluster_rows = FALSE, annotation_row = annot, ...)
amp_patients_idx <- annot$CN > 2.1
map(genes, ~assay(salmon$clean$cvst)[.x,]) %>%
set_names(genes) %>%
map(~wilcox.test(.x[amp_patients_idx], .x[!amp_patients_idx], alternative = "greater")) %>%
map_dbl("p.value")
}
pvals <- plot_region_expr("chr1", "156342801", "157204569", biomart, salmon,
sequenza_1mb_df, scale = "column")