SV Burden
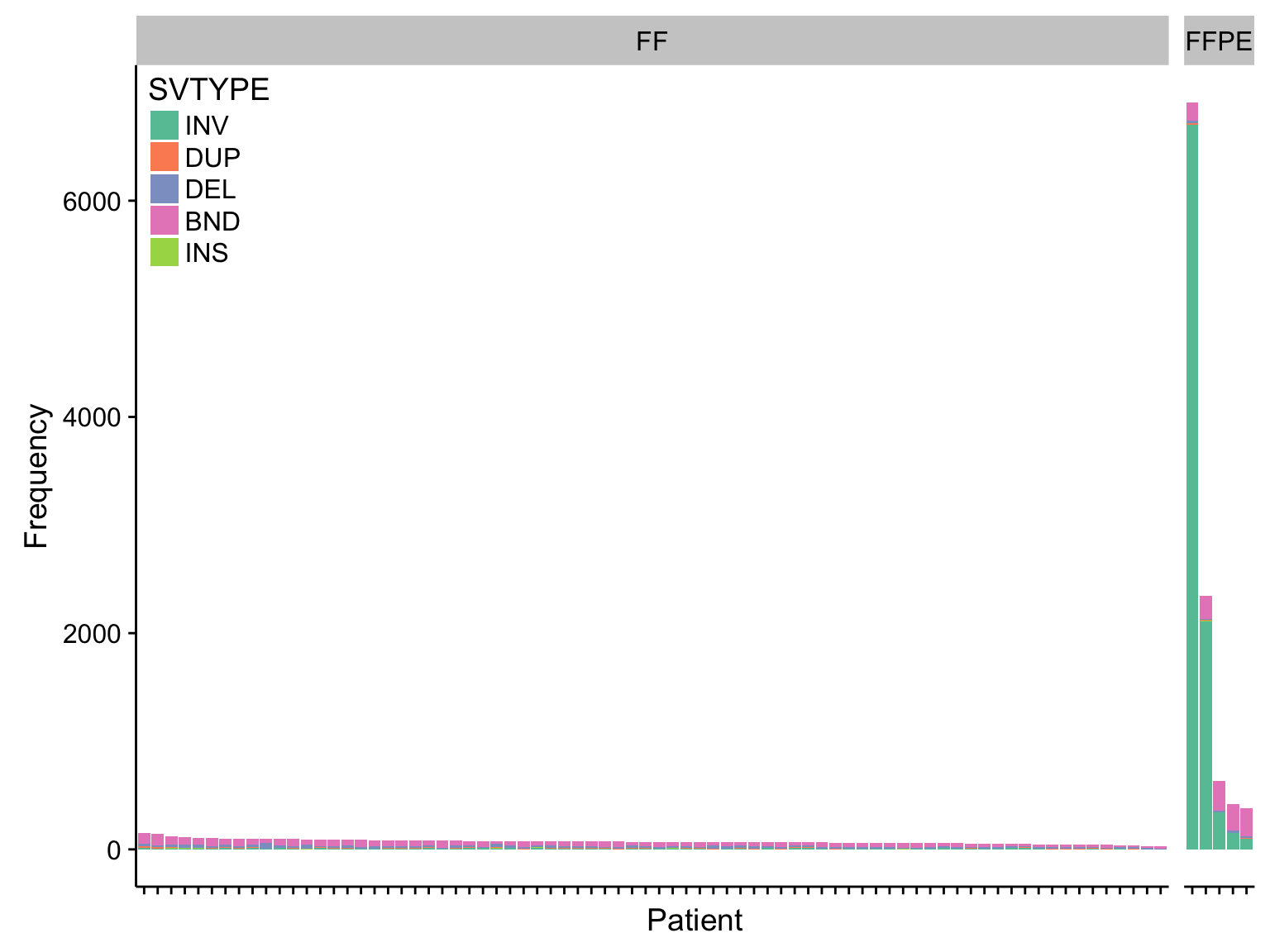
barplot_manta_raw_freq <-
manta_raw %>%
count(patient, ff_or_ffpe, SVTYPE) %>%
ungroup() %>%
mutate(patient = fct_reorder(patient, -n, sum)) %>%
ggplot(aes(patient, n, fill = SVTYPE)) +
geom_col() +
scale_x_discrete(labels = NULL) +
scale_fill_manual(values = c("#66C2A5", "#FC8D62", "#8DA0CB", "#E78AC3", "#A6D854"),
limits = c("INV", "DUP", "DEL", "BND", "INS")) +
facet_grid(~ ff_or_ffpe, scales = "free_x", space = "free_x") +
rotate_x_text() +
place_legend(c(0, 1)) +
labs(x = "Patient", y = "Frequency")
barplot_manta_raw_freq
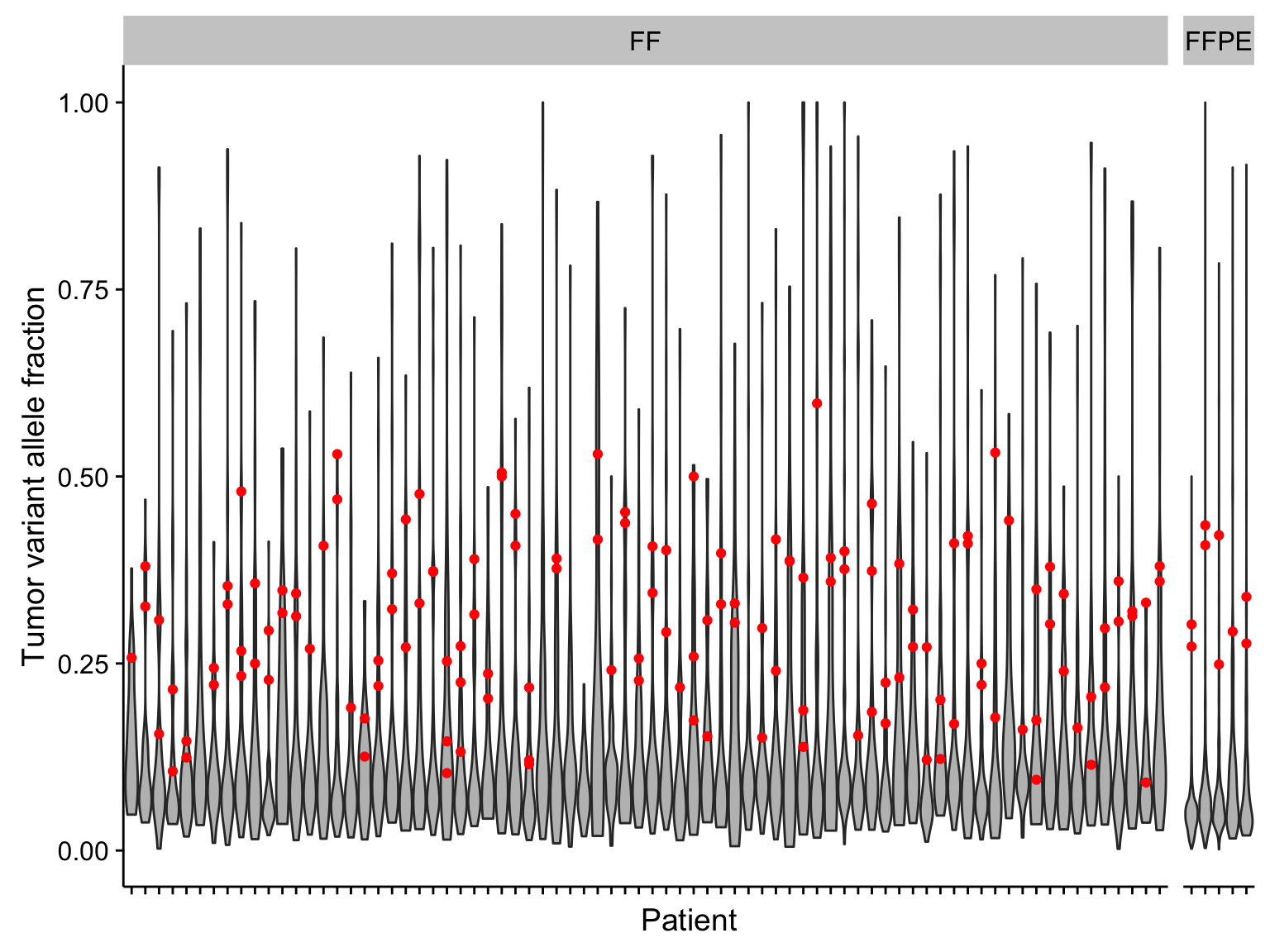
violinplot_manta_raw_vaf <-
manta_raw %>%
mutate(
is_myc = (seqnames == "chr8" & start > 127300000 &
start < 128300000 & grepl("chr(2|14|22)", ALT)),
patient = fct_reorder(patient, -tc)) %>% {
ggplot(., aes(patient, TVAF)) +
geom_violin(fill = "grey", scale = "width") +
geom_point(data = filter(., is_myc), colour = "red") +
facet_grid(~ ff_or_ffpe, scales = "free_x", space = "free_x") +
scale_x_discrete(labels = NULL) +
rotate_x_text() +
labs(x = "Patient", y = "Tumor variant allele fraction")}
violinplot_manta_raw_vaf
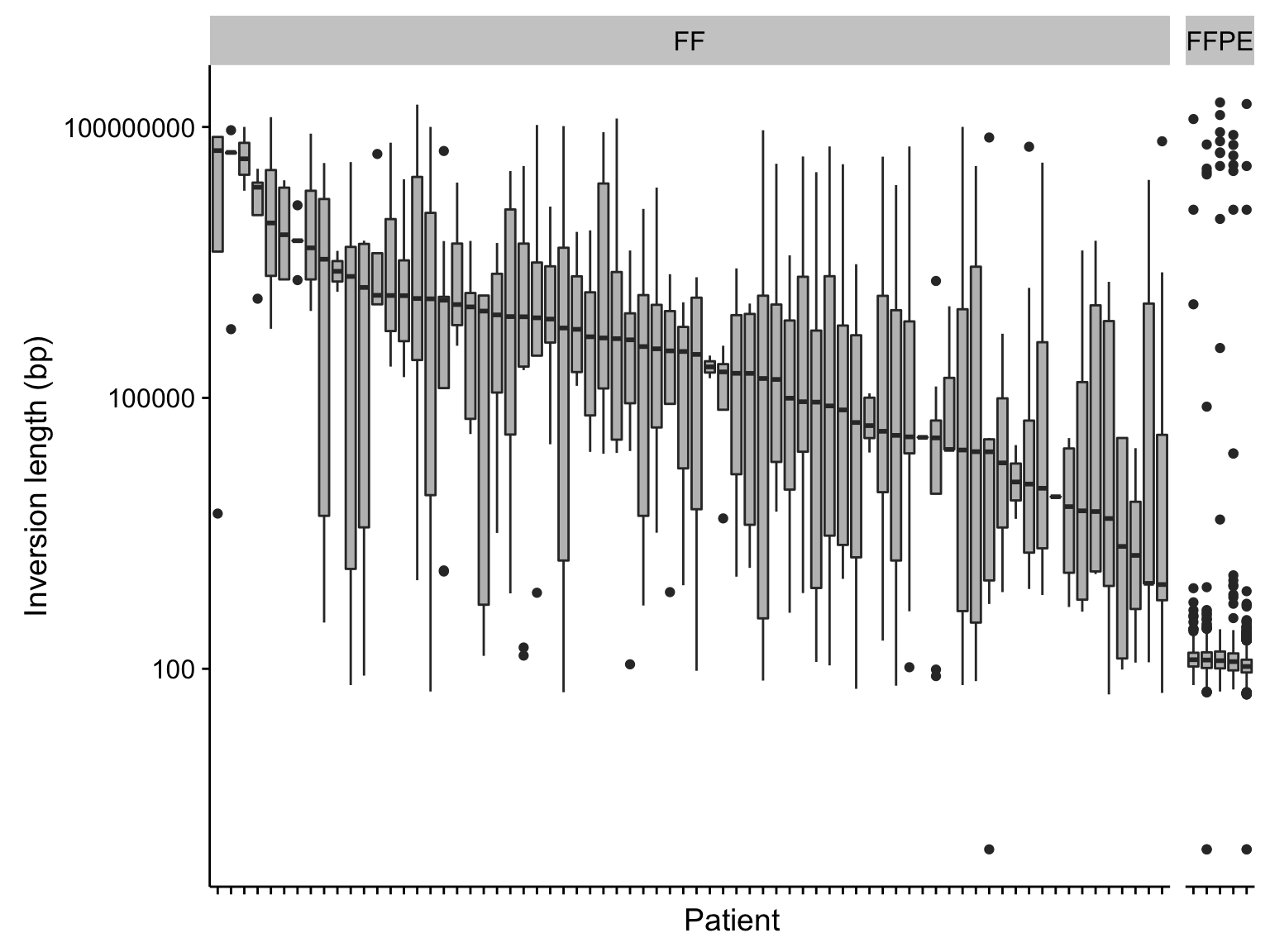
boxplot_manta_raw_inv_svlen <-
manta_raw %>%
filter(SVTYPE == "INV") %>%
mutate(
SVLEN = abs(SVLEN),
patient = fct_reorder(patient, SVLEN, function(x) -median(log10(x)))) %>%
ggplot(aes(patient, SVLEN)) +
geom_boxplot(fill = "grey") +
facet_grid(~ ff_or_ffpe, scales = "free_x", space = "free_x") +
rotate_x_text() +
scale_x_discrete(labels = NULL) +
scale_y_log10() +
labs(x = "Patient", y = "Inversion length (bp)")
boxplot_manta_raw_inv_svlen
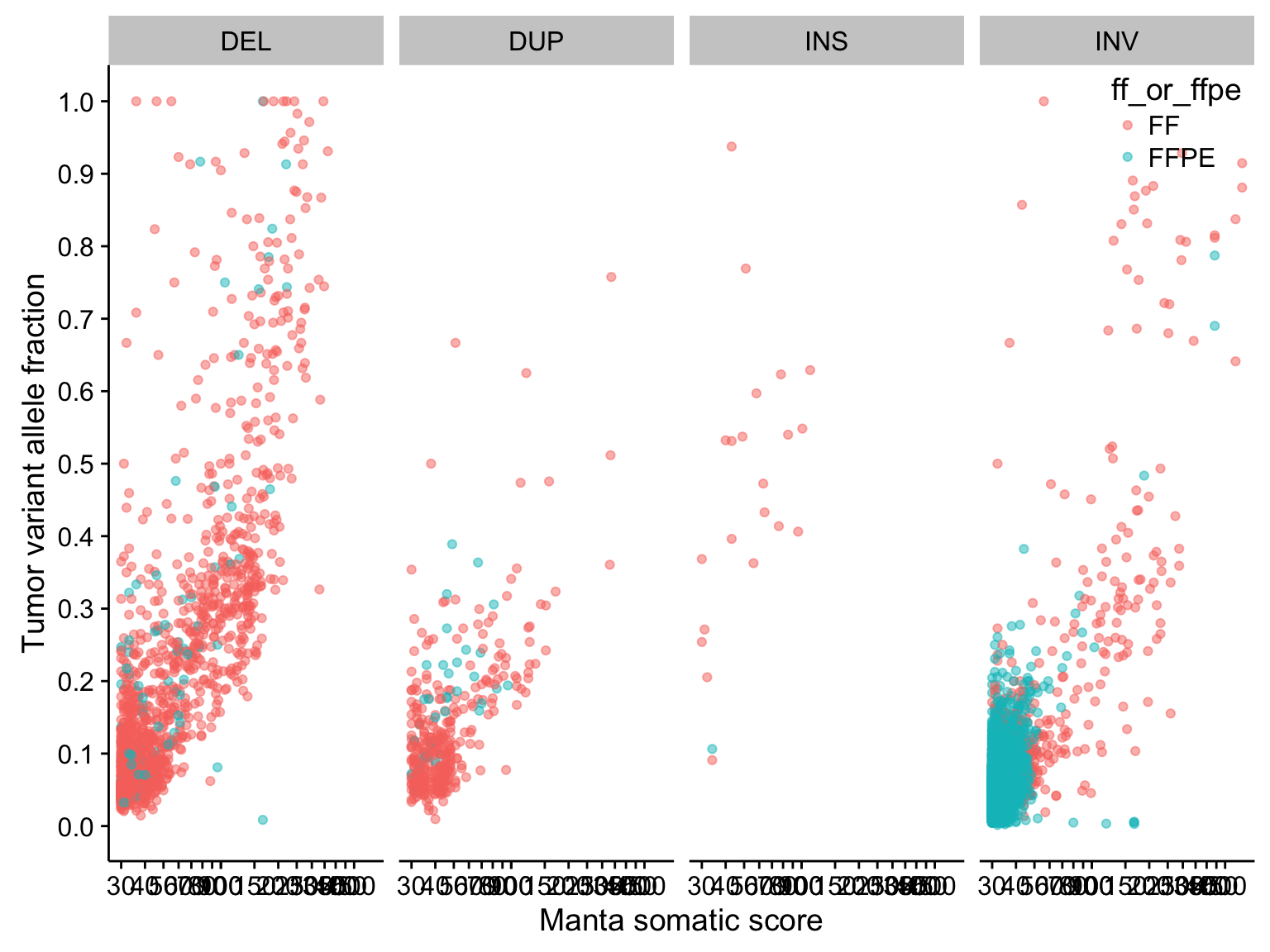
scatterplot_manta_raw_inv_somaticscore_tvaf <-
manta_raw %>%
filter(!is.na(SVLEN)) %>%
mutate(SVLEN = abs(SVLEN)) %>%
ggplot(aes(SOMATICSCORE, TVAF, colour = ff_or_ffpe)) +
geom_point(alpha = 0.5) +
scale_x_log10(breaks = c(seq(0, 100, 10), seq(100, 500, 50))) +
scale_y_continuous(breaks = seq(0, 1, 0.1)) +
facet_grid(~ SVTYPE) +
place_legend(c(1,1)) +
labs(x = "Manta somatic score", y = "Tumor variant allele fraction")
scatterplot_manta_raw_inv_somaticscore_tvaf
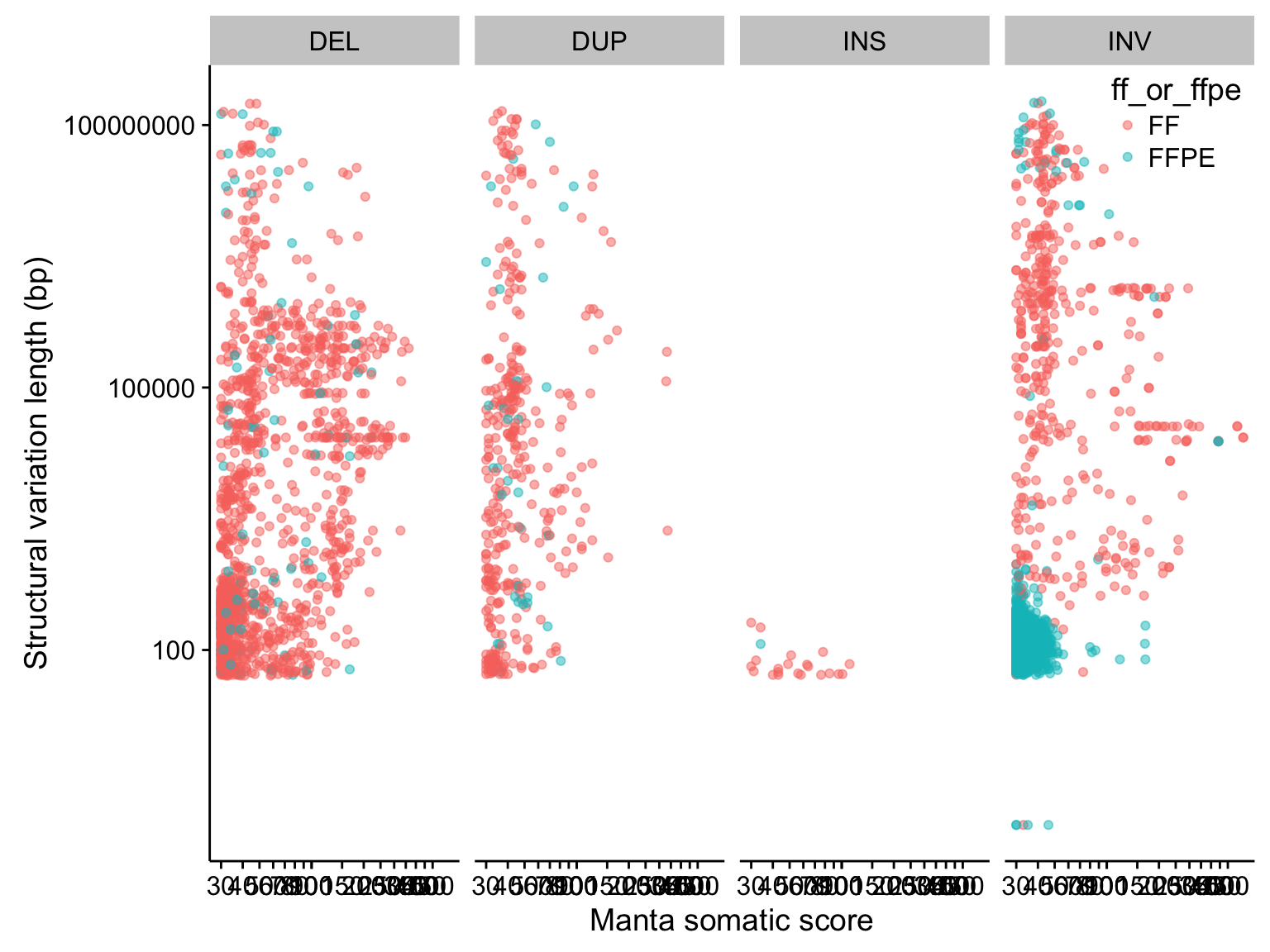
scatterplot_manta_raw_inv_somaticscore_svlen <-
manta_raw %>%
filter(!is.na(SVLEN)) %>%
mutate(SVLEN = abs(SVLEN)) %>%
ggplot(aes(SOMATICSCORE, SVLEN, colour = ff_or_ffpe)) +
geom_point(alpha = 0.5) +
scale_x_log10(breaks = c(seq(0, 100, 10), seq(100, 500, 50))) +
scale_y_log10() +
facet_grid(~ SVTYPE) +
place_legend(c(1,1)) +
labs(x = "Manta somatic score", y = "Structural variation length (bp)")
scatterplot_manta_raw_inv_somaticscore_svlen
scatterplot_manta_raw_bnd_tvaf_somatic_score <-
manta_raw %>%
filter(SVTYPE == "BND") %>%
mutate(is_myc = (seqnames == "chr8" & start > 127300000 &
start < 128300000 & grepl("chr(2|14|22)", ALT))) %>%
group_by(patient, is_myc) %>%
mutate(is_top_myc = ifelse(is_myc, TVAF == max(TVAF), FALSE)) %>% {
ggplot(., aes(SOMATICSCORE, TVAF, colour = is_top_myc)) +
geom_point() +
geom_point(data = filter(., is_top_myc), aes(colour = is_top_myc)) +
scale_color_manual(values = c(`TRUE` = "red", `FALSE` = "grey"), labels = c("Yes", "No")) +
scale_x_log10(breaks = c(seq(0, 100, 10), seq(100, 400, 50))) +
scale_y_continuous(breaks = seq(0, 1, 0.1)) +
place_legend(c(1,1)) +
labs(x = "Patient", y = "Tumor variant allele fraction", colour = "MYC translocation")}
scatterplot_manta_raw_bnd_tvaf_somatic_score

manta <- filter(manta_raw, TVAF > 0.1, SOMATICSCORE > 50)